Определение. Заболевание Х-сцепленное доминантное, поражающее кожу, ЦНС, глаза и костную систему и характеризующееся на определенной стадии своего развития типичной меланиновой пигментацией.
Историческая справка. В 1906 г. А. Е. Garrod (1857—1936) написал отчет об умственно отсталой девочке с тетраплегией и характерной пигментацией кожи. В 1926 г. швейцарский дерматолог Бруно Блох (1878—1933) описал случай данного заболевания и первым предложил термин «недержание пигмента». Более детально изучил заболевание американский дерматолог профессор Марион Сульцбергер (1895-1983) в 1927 г. Он описал своеобразные эктодермальные и мезодермальные дефекты, которые возникают в младенческом возрасте и проявляются буллезной сыпью, а в дальнейшем характерной пигментацией кожи. В 1929 г. появилась еще одна публикация, которую выполнил H.W.Siemens. В 1989 г. A. Sefiani с соавторами выявили локализацию гена в хромосоме Xq28 при данном синдроме. В 1993 г. была выполнена публикация S.J. Landy и D. Donnai с обширным обзором случаев с синдромом Блоха—Сульцбергера.
Этиология и патогенез. Генетическое заболевание наследуется доминантно и сцеплено с полом. Характеризуется нарушением меланогенеза. Развитие пигментации связано с тем, что имеется функциональная аномалия пигментообразующих клеток базального слоя эпидермиса. Из-за патологической проницаемости клеточной мембраны для меланина эти клетки отдают в дерму весь или почти весь вырабатываемый ими меланин. Линейные образования на коже при данном синдроме обусловлены мозаицизмом вследствие инактивации Х-хромосомы. Были выявлены два генетических локуса при этом синдроме. При I типе ген локализуется в хромосоме Xq11, болезнь характеризуется спорадическими случаями, генетические аномалии возникают без предшествующей воспалительной фазы. При II типе (семейный вариант заболевания) ген расположен в хромосоме Xq28. При этом типе синдрома отмечаются Х-сцепленные доминантные мутации в гене NEMO.
Предполагают, что эти мутации приводят к снижению иммунной толерантности в эктодермальных тканях. У гетерозиготных девочек это вызывает реакцию, похожую на аутоиммунную, а у гомозиготных мальчиков — реакцию по типу «трансплантат против хозяина», приводящую к фатальному исходу заболевания. Развитию заболевания способствуют внутриутробные травматизация, аллергизация и инфекция плода, нейрогуморальные нарушения. Возраст и пол. В 95—97% случаев регистрируется только у лиц женского пола. Плод мужского пола обычно погибает внутриутробно. В мировой литературе отмечено выживание мальчиков в 30 случаях. Дети уже рождаются с клиническими проявлениями или заболевание начинается в течение первых 6 недель жизни. Поражения кожи. В течении синдрома выделяют четыре стадии. Первая является воспалительной (везикулярной).
Длится от момента рождения и в течение нескольких первых месяцев жизни. Имеются эритематозные, уртикарные, пузырьковые и пузырные элементы на коже туловища и (преимущественно) конечностей. Лицо, ладони, подошвы и слизистые не поражаются.
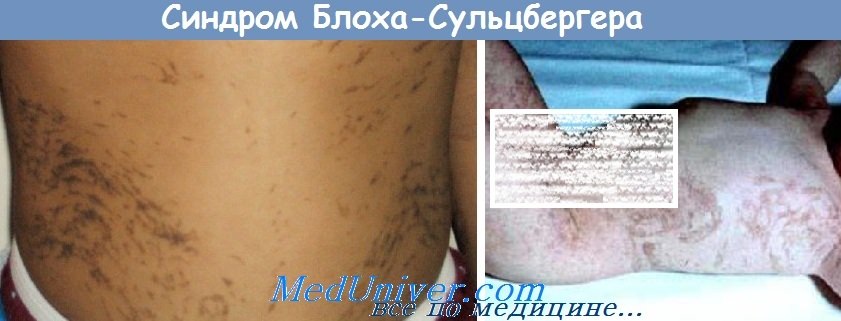
Высыпания имеют тенденцию к полосовидному, вихревидному расположению. Вторая стадия называется веррукозной и длится до первого года жизни. На тех же участках появляются лихеноидные, лентикулярные папулы и веррукозные высыпания. На 3—6-м месяце жизни папулы постепенно рассасываются. Вторая стадия отмечается у 2/3 больных. Для третьей стадии характерны гиперпигментации.
Длится с одного года до подросткового возраста. Эта стадия присутствует у всех пациентов с синдромом Блоха-Сульцбергера. На фоне папулезной сыпи или в период ее разрешения появляются пигментные пятна от темно-серого до темно-коричневого цвета, иногда с ливедо-синеватым оттенком. Гиперпигментации имеют полосовидную, сетчатую, паукообразную форму или в виде завихрений. Очаги располагаются по линиям Блашко (но не по дерматомам или линиям расщепления кожи). Гиперпигментации локализуются на голове, туловище и конечностях, часто напоминают брызги. Примерно у 40% больных возникновение пигментации не связано с предшествующими высыпаниями. Для четвертой стадии характерны гипопигментации. Между периодом полового созревания и 25—30 годами гиперпигментации спонтанно исчезают, оставляя легкую атрофию с гипопигментацией. В этих зонах отсутствуют волосы и потовые железы. Такие изменения отмечают преимущественно на нижних конечностях.
Другие изменения кожи и ее придатков. В части случаев у больных обнаруживают очаги рубцовой алопеции на волосистой части головы, иногда дистрофию ногтей (около 10% случаев), диффузную алопецию.
Другие изменения. Легочная гипертензия, добавочные соски, врожденные пороки сердца, отсутствие или поражение зубов, преимущественно премоляров и верхних боковых резцов (замедление прорезывания, патология дентина, коническая форма). Во время воспалительной фазы в крови и пузырной жидкости выявляют эозинофилию (50—74%), лейкоцитоз периферической крови.
Гистология. В I стадии наблюдают спонгиоз с образованием пузырьков и пузырей, содержащих эозинофилы, фибрин. Местами рассеяны крупные дискератотические клетки с гомогенной протоплазмой. Дерма инфильтрирована мононуклеарами и эозинофилами.
Во II стадии картина может быть сходна с псевдоэпителиоматозной гиперплазией. Отмечаются акантоз, папилломатоз и гиперкератоз с внутриэпидермальной кератинизацией (кератиноциты располагаются спирально), местами — вакуольная дистрофия базальных клеток. В дерме — отек и инфильтраты из гистиоцитов, лимфоцитов, нейтрофильных и эозинофильных гранулоцитов, много меланофоров. В III стадии воспалительные явления уменьшаются или исчезают. В верхних частях собственно кожи, главным образом в сосочковом слое, отмечается обильное скопление пигмента. Заполненные пигментом соединительнотканные клетки так густо лежат на самой дермоэпидермальной границе, что получается впечатление их «контакта» с базальными клетками. В клетках базального слоя меланина мало или содержание его в пределах нормы, в них отмечается вакуолизация. Выявляются зоны истончения эпидермиса, очаговый гиперкератоз. В IV стадии — снижение количества меланина, фиброзные изменения и отсутствие или снижение придатков кожи в пораженных областях.
Диагноз ставят по клинической картине кожных поражений в зависимости от стадии заболевания, а также на основании результатов инструментальных и генетических исследований.
Дифференцируют на воспалительной стадии с герпесом, буллезными дерматозами: эпидермолизом, линейным IgA и пемфигоидом. На веррукозной стадии дифференцируют с эпидермальным невусом, на стадии гиперпигментации — с линейным и завитым невоидным гипермеланозом, на стадии гипопигментации — с гипомеланозом Ито.
Течение и прогноз. У 60—80% пациентов с этим синдромом имеются системные аномалии экто- и мезодермального происхождения. При возникновении припадков на первой неделе жизни прогноз сомнительный, отмечается задержка развития. Отсутствие припадков и соответствие развития возрастным нормам свидетельствуют о благоприятном прогнозе. Пигментации персистируют на протяжении многих лет и постепенно исчезают в подростковом или раннем взрослом возрасте.
Лечение. В воспалительной стадии в виде наружного лечения используют глюкокортикоиды. При образовании пузырьков накладывают повязки, смоченные антисептическим раствором (бледно-розовый раствор калия перманганата). При выраженных воспалительных изменениях назначают глюкокортикоиды внутрь, например преднизолон 0,5—1,0 мг/кг в день с быстрой отменой. Проводят профилактику вторичной инфекции. Поражения других органов лечат у смежных специалистов.