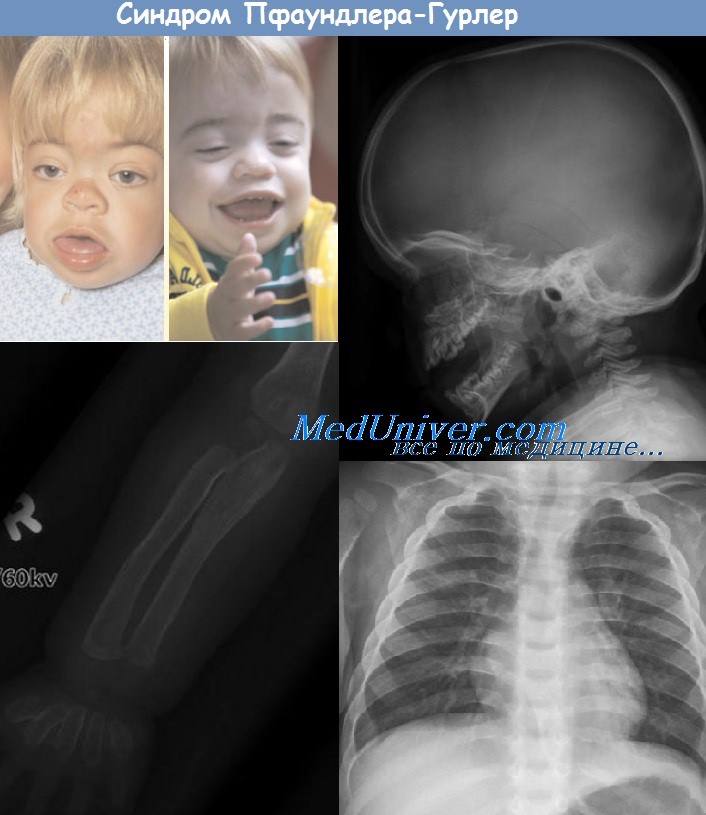
Мукополисахаридоз I типа (болезнь Гурлер)—наследование аутосомно-рецессивное. В клетках и межклеточном веществе откладываются дерматапсульфат, гепарансульфат и ганглиозиды. Имеется дефицит фермента бета-галактозидазы и, по последним данным, а-идуронидазы.
Отмечаются тяжелые симптомы гаргоилизма с прогрессирующими нарушениями психики, ранним помутнением роговицы, глухотой, гепато- и спленомегалией. В крови лимфоциты с гранулами, обладающими метахромазией, в моче резко повышено содержание мукополисахаридов. Дети умирают в возрасте 10—12 лет при явлениях поражения головного мозга (гидроцефалия), сердца или от интеркуррентных инфекционных заболеваний.
Myкополисахаридоз II типа (болезнь Гэнтера)—наследование рецессивное, связанное с Х-хромосомон, В тканях откладываются дерматансульфат и гепарансульфат, имеется дефицит |3-галактозидазы и, по последним данным, отсутствие фермента а-идуроносульфат сульфатазы [Swift Th., Mac Donald Th., 1976]. Заболевание течет длительно по сравнению с болезнью Гурлера, симптоматика не так резко выражена, отсутствует помутнение роговицы. В моче количество мукополисахаридов повышено, в лимфоцитах крови — гранулы, обладающие метахромазией.
Myкополисахаридоз III типа (болезнь Санфилиппо) — наследование аутосомно-рецессивное. В тканях накапливается гепарансульфат. Имеется дефицит бета-галактозидазы. Кроме того, в органах снижена активность гепарансульфатазы [Cain H. et al., 1976]. Над соматическими изменениями преобладает задержка психомоторного развития. Однако, по данным Н. Cain и соавт. (1976), содержание ГАГ в печени увеличено в 12,5 раз против нормы, в основном за счет накопления гепарансульфата. Аналогичное вещество обнаруживается в нейронах головного мозга, в стенках артерий, селезенке. В моче количество ГАГ повышено, в лимфоцитах крови — метахроматические гранулы.
Мукополисахаридоз IV типа (болезнь Моркио)—наследование аутосомно-рецессивное. В тканях откладывается кератаисульфат вследствие дефицита N-ацетилгексозаминсульфатазы. Преобладают изменения скелета и помутнение роговицы. Гранулы в лимфоцитах отсутствуют.
Myкополисахаридоз V типа (болезнь Шейе)—в настоящее время рассматривается как вариант мукополисахаридоза I типа, проявляющегося клинически у взрослых, так как активность фермента а, L-идуронидазы снижена незначительно.
Мукополисахаридоз VI типа (болезнь Марото — Лами) — наследование аутосомно-рецессивное.
В тканях откладывается дерматансульфат. Имеется дефицит фермента 4-ацетилгалактозамин-4-сульфатазы. В остальном болезнь сходна с мукополисахаридозом I типа, однако нарушения психомоторного развития очень незначительны или совсем отсутствуют.
Мукополисахаридоз VII типа характеризуется недостаточностью фермента бета-глюкуронидазы. В тканях накапливается и выделяется с мочой дерматансульфат. Проявления болезни сходны с мукополисахаридозом I типа, однако не так резко выражены.
Мукополисахаридозы диагностируются по типичным клиническим симптомам, а также на основании биохимического определения ГАГ в моче и наличия метахроматических гранул в лимфоцитах крови. Гистохимическое изучение затруднено, так как ГАГ чрезвычайно легко растворяются при фиксации тканей, а морфологические изменения малохарактерны. Если заболевание встречается в семьях, рекомендуется амниоцентез, исследование амниотической жидкости и культуры клеток амниона на присутствие ГАГ.
Итак, в заключение следует указать, что, несмотря на преобладающие нарушения обмена ГАГ при так называемых «чистых» мукополисахаридозах, они фактически представляют собой группу заболеваний, при которых в большинстве случаев имеются нарушения смешанного типа— мукополисахаридов и ганглиозидов.