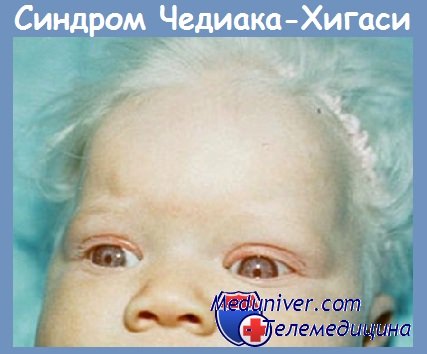
Синдром Чедиака — Хигаси — редкое заболевание, характеризующееся рецидивирующими пиогенными инфекциями, окулокутанным альбинизмом и наличием гигантских цитоплазматических гранул во многих клетках, преимущественно в лейкоцитах периферической крови. Болезнь наследуется аутосомно-рецессивно. Причину пиогенных инфекций связывают с нарушением бактерицидной активности лейкоцитов и нейтропенией.
Снижение бактерицидной активности зависит ют дефекта лизосом, а также от снижения способности лейкоцитов к хемотаксису. Кроме того, полагают, что фагоциты больных обладают повышенной активностью к аутофагоцитозу, что приводит к гиперспленизму и вторичной нейтропении. Дефект пигментации зависит от нарушений распределения меланофоров и аномального расположения меланосом.
У больных наблюдается гипопигментация кожи, радужной оболочки глаза и волос, повышенная чувствительность кожи к солнечному свету. Имеется, вероятно, связь нарушений пигментации с дефектом фагоцитарной системы. Течение инфекционных процессов также неблагоприятное, как и при ХГБ, наблюдаются они с периода новорожденности. Чаще, чем при ХГБ, поражается желудочно-кишечный тракт.
Диагноз синдрома подтверждается наличием гигантских включений в лейкоцитах, дающих положительную реакцию на пероксидазу.
Синдром Джоба характеризуется развитием рецидивирующих «холодных» абсцессов, вызванных золотистым стафилококком, встречается у девочек с нежной белой кожей и белокурыми или рыжими волосами. Некоторые считают его вариантом ХГБ. Предполагается аутосомно-рецессивная наследственность. А. Т. Devis и соавт., впервые подробно описавшие этог синдром в 1966 г., считали, что он является следствием дефекта местной резистентности ткани к стафилококку. Изучение киллерной способности лейкоцитов дает противоречивые результаты.
По одним данным, имеется дефект гексозомоносфатного шунта, как при ХГБ, по другим — киллерная способность лейкоцитов не нарушена [Quic P. О., Devis А. Т., 1973]. Абсцессы располагаются в коже, подкожной клетчатке, лимфатических узлах, легких, печени. Могут наблюдаться экзема, хронический ринит, отит. Содержание иммуноглобулинов нормальное, отмечается только повышение содержания IgE.
Абсцессы характеризуются отсутствием реактивных изменений со стороны окружающей ткани (отсюда их название «холодные» абсцессы), нет гиперемии, лейкоцитарного воспалительного вала. Данные биопсии кожи указывают на наличие вокруг абсцессов большого количества эозинофилов и базофилов при резком снижении количества нейтрофильных лейкоцитов.
Таким образом, у больных имеется отчетливое снижение резистентности к стафилококковой инфекции и нормальная способность к реакциям специфического клеточного и гуморального иммунитета, увеличение количества IgE и эозинофилия. Этиологическая и патогенетическая связь между всеми перечисленными изменениями неясна и требует дальнейшего изучения.
Дефект лейкоцитарного хемотаксиса Miller и соавт. в 1971 г., А. Соnstantopoules и соавт. в 1978 г. обнаружили у двух детей, у которых он сопровождался повторными тяжело текущими инфекциями и нейтропенией. Лейкоциты имели нормальную бактерицидную способность, но не были способны к хемотаксису in vitro. При этом количество лейкоцитов в костном мозге было нормальным, в периферической крови была стойкая нейтропения.
Окончатая проба с воспалением показывала появление миграции лейкоцитов только после инфузии свежей плазмы.
Наследственный детский агранулоцитоз (агранулоцитоз Костмана)—заболевание грудных детей, характеризующееся агранулоцитозом периферической крови и гипоплазией миелопоэза костного мозга. Впервые описан шведским педиатром Костманом в 1956 г. Характеризуется лихорадкой, кожными воспалительными поражениями и симптомами общего инфекционного заболевания. В периферической крови — нейтропения вплоть до полного исчезновения зернистых лейкоцитов. Прогноз неблагоприятный.
Этиология: аутосомно-рецессивная наследственность, возможно, сцепленная с Х-хромосомой, болеют мальчики. Заболевание описывается в семьях, где имеет место кровное родство родителей. Патогенез не установлен.
В костном мозге полное отсутствие или резкая степень торможения пролиферации гранулоцитарного ростка при сохранности моноцито-, и эритропоэза. В тимусе преждевременный жировой метаморфоз. В коже, в мягких тканях и по органам (в частности в легких) некрозы с микробными эмболами без лейкоцитарной реакции и без нагноения. Смерть наступает от сепсиса в первые месяцы жизни.